plexDIA optimization
Data-Driven Optimization of DIA Mass Spectrometry by DO-MS
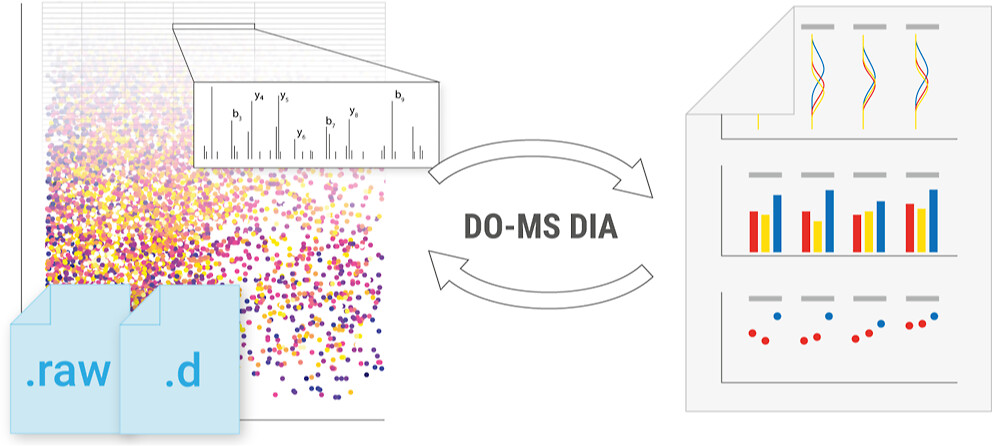
Mass spectrometry (MS) enables specific and accurate quantification of proteins with ever-increasing throughput and sensitivity. Maximizing this potential of MS requires optimizing data acquisition parameters and performing efficient quality control for large datasets. To facilitate these objectives for data-independent acquisition (DIA), we developed a second version of our framework for data-driven optimization of MS methods (DO-MS). The DO-MS app v2.0 (do-ms.slavovlab.net) allows one to optimize and evaluate results from both label-free and multiplexed DIA (plexDIA) and supports optimizations particularly relevant to single-cell proteomics. We demonstrate multiple use cases, including optimization of duty cycle methods, peptide separation, number of survey scans per duty cycle, and quality control of single-cell plexDIA data. DO-MS allows for interactive data display and generation of extensive reports, including publication of quality figures that can be easily shared. The source code is available at github.com/SlavovLab/DO-MS.
Wallmann G., Leduc A., Slavov N., Data-Driven Optimization of DIA Mass Spectrometry by DO-MS, J. Proteome Res., doi: 10.1021/acs.jproteome.3c00177; Preprint at bioRxiv doi: 10.1101/2023.02.02.526809, PDF